This post picks up from an earlier introduction to the anti-apoptotic Bcl-2 protein and an FDA approved drug (venetoclax), a selective inhibitor of Bcl-2. In brief, we summarized why Bcl-2 is an attractive target for therapeutic development and highlighted how venetoclax binds to a large pocket on the protein.
Here we will (re)dock and cross-dock the ligand from a co-crystal structure to Bcl-2 using Autodock Vina1.
A related software package2 (smina) is built upon the original Autodock Vina and has some additional scoring and minimization features, which will be covered in a later post.
While there are limitations to the usefulness of re-docking3, it is a relatively simple exercise to do when first starting with a new model and it can sometimes reveal interesting information about the system (and serves as a quick sanity check) prior to cross-docking or virtual screening. Cross-docking - whereby you extract the ligand from a co-crystal structure and dock to a different, but related structure is typically a better challenge to evaluate docking robustness. Minor conformational differences (especially in sidechain packing or rotamers) are not uncommon and can have a large impact on the docking poses.
See below for some opinionated notes on model validation.
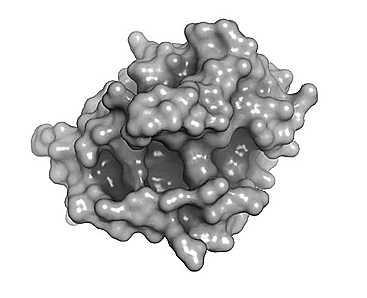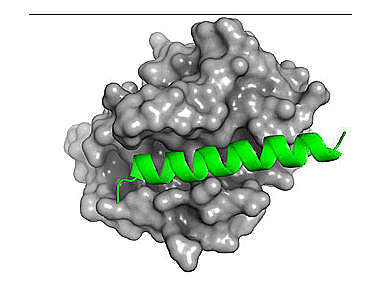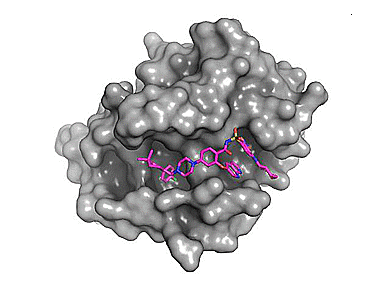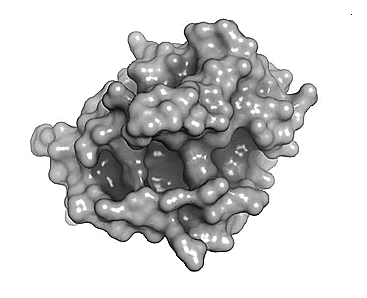
Figure 1. Comparison of the binding site for the small molecule inhibitor, venetoclax (magenta) and a native ligand - BH3 peptide (green). PDB: 6O0P and 2XA0, respectively.
We see above that the small molecule inhibitor, venetoclax, binds to a long trench or groove in the structure of Bcl-2. This is the same pocket that facilitates binding of Bcl-2 to its protein binding partners (via homo- or hetero-dimerization).
Autodock Vina is an open source molecular docking program developed at the Scripps Research Institute by Dr. Oleg Trott1. There are many docking programs (both free and commercial) available including: Glide, MOE, GOLD, rDock, DOCK, and many more. With over 60+ docking programs available, everyone has their favorite and numerous reviews comparing the different packages have been published4,5.
Using Autodock Vina, re-docking venetoclax to the Bcl-2 G101A mutant construct (PDB: 6O0P) yields a series of possible binding modes (cyan), the first of which very closely resembles the binding pose of the co-crystallized ligand (magenta).
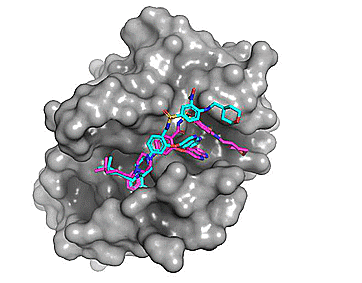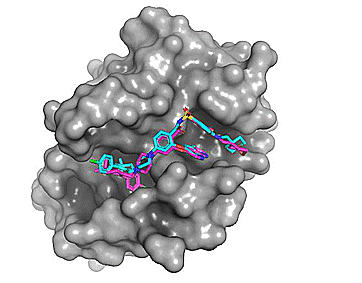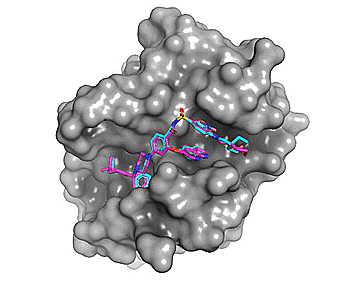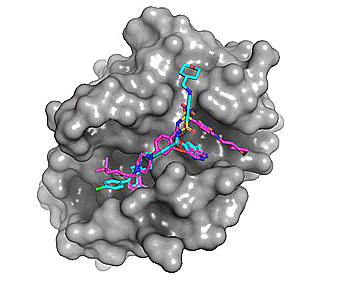
Figure 2. Top 8 binding poses for venetoclax (re)docked to Bcl-2 G101A (PDB: 6O0P)
Cross-docking the venetoclax ligand (from PDB: 6O0P) to a related inhibitor-bound structure of WT Bcl-2 (4IEH) yields docking poses that deviate more from the true binding mode than do the re-docked poses. However, overall the lowest energy pose from the cross-docking still closely resembles the true binding mode found in the co-crystal (see pose mid-animation).
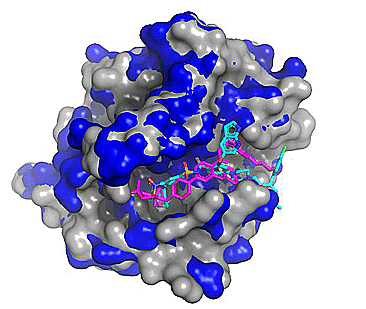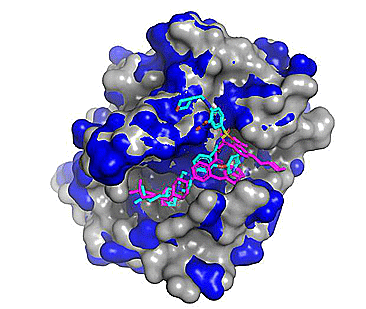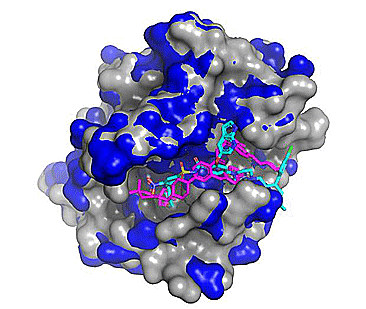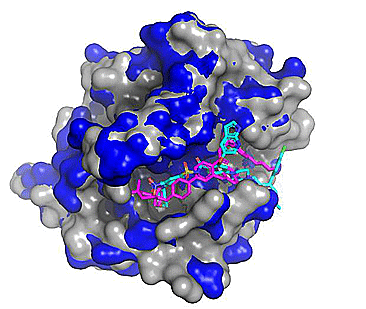
Figure 3. Top 5 binding poses for venetoclax (from PDB: 6O0P; Protein - blue, Co-crystallized ligand - magenta) cross-docked to WT Bcl-2 (PDB: 4IEH; protein - grey, Cross-docked poses - cyan). Note the regions where the blue and grey protein surfaces differ as these correspond to conformational differences (mostly side chain) between the two related protein structures.
Model validation:
This is a critical part of any virtual screening / docking workflow. The takeaway is that the model should have predictive power and should therefore always be validated against experimental data. Model preparation, minimization, and optimization can be done exhaustively (and by following stringent protocols, best practices, etc) only to find out that has poor predictive power. A well-ordered (highly structured) binding pocket lacking significant conformational exchange might be a far superior model (in terms of predictive power) than even the most exhaustively "prepared" model of a highly dynamic, disordered pocket.
A note on dynamics:
It is a common error to assume a static picture of proteins and their putative binding pockets - an error which can in part be attributed by the fact that x-ray structures present us with a static view of the target (unless we consciously take this into account via B-factor, NMR restraints (NOE), compare to other related structures, simulate dynamics via MD, etc). All too often can a lack of consideration for the dynamic nature of proteins result in misleading rational design efforts!
References
1. Journal of Computational Chemistry 31 (2010) 455-461, http://vina.scripps.edu/
2. https://sourceforge.net/projects/smina/files/
3. https://www.cheminformania.com/never-use-re-docking-for-estimation-of-docking-accuracy/
4. Biophys Rev. 2017 Apr; 9(2): 91–102.
5. Phys. Chem. Chem. Phys., 2016,18, 12964-12975